

X-linked sideroblastic anemia (XLSA) is an anemic disease caused by mutations in the gene encoding 5-aminolevulinate synthase 2 (ALAS2), which catalyzes the rate-limiting step of heme biosynthesis. With current interventions including pyridoxine treatment and allogeneic hematopoietic stem cell transplantation, severe unmet needs remain for patients with XLSA. Here, we used CRISPR/Cas9 technology to efficiently and functionally correct the pathogenic mutation in intron 1 of ALAS2 in CD34+ hematopoietic stem cell and progenitor cells (HSPCs) from two patients. Intriguingly, we found that the gene-editing efficiency of CD34+HSPCs from the elder patient was much lower than that from the younger patient, consistent with the poor hematopoiesis of older XLSA patients observed in clinical practice. Furthermore, we performed single cell RNA-sequencing (scRNA-seq) to investigate the causes of those phenomenon in-depth.
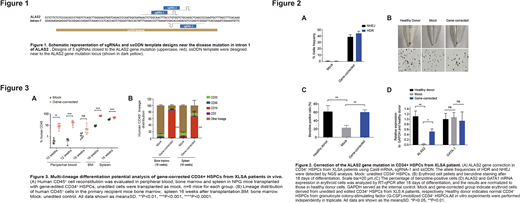
Previously we and others identified the A>G mutation in the GATA1 binding region of intron 1 of ALAS2 in particular XLSA families and have demonstrated the key role of this site in regulating ALAS expression. Thus, we first designed a series of sgRNAs, along with single-stranded DNA oligonucleotide donors (ssODN). After co-electroporating with Cas9 mRNA into patient-derived hiPSCs, sgRNA-1 and ssODN were selected for further experiments based on optimal correction rate (43.93±3.43% via HDR). Next, using a well-established erythroid protocol, CD34+ HSPCs of control and gene-edited groups were differentiated into erythroid cells in vitro. Surprisingly, heme biosynthesis examined by benzidine staining showed that compared with the mock cells, the gene-corrected group significantly increased the frequency of benzidine-positive cells.
To examine the multilineage differentiation potential of gene-corrected CD34+ HSCPs, we performed colony-forming unit (CFU) assays to quantify various types of colonies. Compared with mock cells, gene-edited group significantly enhanced the generation of total, CFU-GM and BFU-E colonies, suggesting higher clonogenic potential. Next, gene-corrected CD34+ HSPCs were transplanted into nonobese diabetic (NOD)/Prkdcscid/IL-2Rγnull (NPG) mice to evaluate the repopulating potential. All transplanted mice displayed engraftment in multiple organs at 10-16 weeks post transplantation, and the gene-corrected cells showed greater engraftment potential than mock group. In addition, hematopoietic reconstitution analysis indicated that the gene-corrected cells maintained normal lineage distribution, while the B cell development of mock group was impaired. Moreover, gene-editing efficiency analysis of bone marrow samples 16 weeks after transplantation exhibited high editing rate (34±7.18% via HDR), comparable to the in vitro efficiency. The specificity of the Cas9 mRNA-based gene editing system was examined using unbiased Digenome-Seq. In total, 32 potential off-target sites were identified and deeply interrogated via targeted PCR and NGS analysis of XLSA iPSCs electroporated with Cas9 mRNA and sgRNA. No off-target cleavage events were detected at these sites, suggesting a lack of detectable off-target events.
Finally, scRNA-seq of CD34+ HSPCs from healthy donor and XLSA patients revealed more HSC/LMPP and erythroid progenitor cells in older XLSA patient. Further analysis showed that cell cycle and gene expression in older HSC/LMPP cells were significantly different from that from healthy donors and younger patients. Hence, we speculated that the compensatory differentiation of HSCs caused by long-term functional red blood cell deficiency caused the abnormal expansion of HSCs, which led to the poor hematopoiesis in elderly patients.
Our study firstly uses CRISPR/Cas9 gene-editing technology to correct the disease mutation in patient's CD34+ HSPCs and rescues ALAS2 expression and heme biosynthesis, directly confirming that this mutation is the pathogenic factor for XLSA. In addition, we dissect the transcriptional profile of CD34+ HSCPs from XLSA patients at single cell resolution for the first time, shedding light on mechanistic insights into the XLSA pathogenesis. The robust gene-correction rates and significant function rescue in patient's CD34+ HSPCs further suggest a curable option of gene-edited HSC transplantation for the treatment of the patients with XLSA.
Fang:EdiGene Inc.: Current Employment. Yuan:EdiGene Inc.: Current Employment. Yang:EdiGene Inc.: Current Employment. Yu:EdiGene Inc.: Current Employment. Zhang:EdiGene Inc.: Current Employment. Shi:EdiGene Guangzhou Inc.: Current Employment. Qi:Novogene Co, Ltd: Current Employment. Wei:EdiGene Inc.: Current Employment.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal